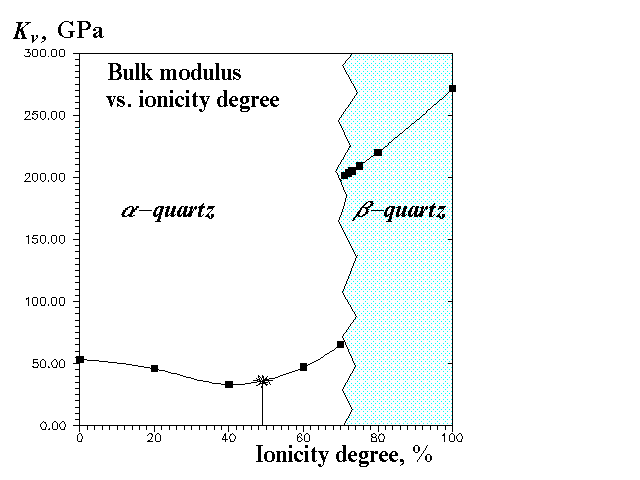
THEORETICAL INVESTIGATION OF a--b TRANSFORMATION OF QUARTZ
Artem R.Oganov, Vadim S. Urusov, Nickolai N. Eremin.
Dept. of Crystallography and Crystal
Chemistry, Moscow State University, 119899, Moscow, Russia.
E-mail : oganov@openmail.irex.ru
Keywords: quartz, computer simulation, phase transition, atomization energy.
The atomization energy minimization method [1,2] has been applied for calculation (METAPOCS code) of dependencies of crystal structure, elastic properties and atomization energy of quartz (SiO2) on the ionicity degree of Si-O bonds - f(Si-O) (for details see [1]).
Our interatomic potential includes Coulombic interactions, Born-Mayer repulsion, van der Waals attraction, O-Si-O and Si-O-Si three-body bond-bending terms; covalent terms in the interaction energy were imitated by Morse potentials. Ionic and covalent contributions were weighted in accordance to f(Si-O). The atomization energy is obtained from the lattice energy, corresponding to variable atomic charges (bond ionicities), via its correction to the charge-transfer energy; the latter being a work, required for a partial ionization of an atom. Some potential parameters were taken from the literature, others were chosen using atomic and molecular constants; the others being obtained from fitting of the minimum-energy crystal structure, atomization energy and elastic properties to the experimental ones.
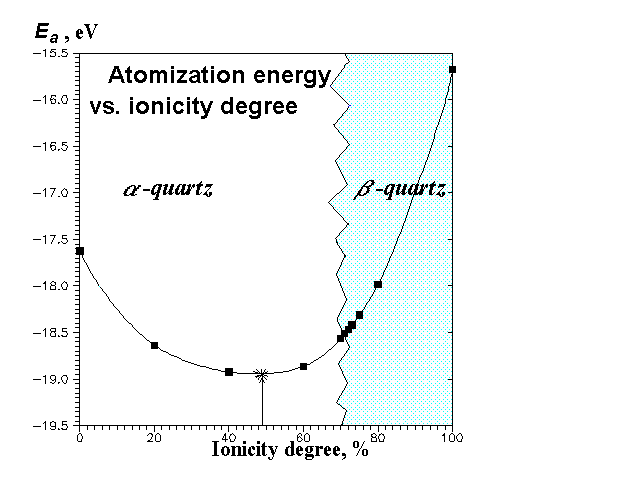
With the parameters chosen the atomization energy has a minimum at f(Si-O)=48% (Figure 1); its value at this ionicity degree (-18,96 eV) closely corresponds to the experimental one (-19,47 eV). Description of the crystal structure (see Table) and elastic properties at this ionicity degree is very good.
Table. Crystal structure of -quartz : theory and experiment.
Lattice parameters |
Calculation
|
Experiment [3] |
a0, A |
4.8928 |
4.91239 |
c0, A |
5.3542 |
5.40385 |
Vo, A3 |
111.01 |
112.933 |
Space group P3121 (Z=2) |
||
Atomic coordinates |
Calculation (f(Si-O)=48%). |
Experiment [3] |
Si |
(0.4634; 0; 1/3) |
(0.4701; 0; 1/3) |
O |
(0.4240; 0.2756; 0.2139) |
(0.4139; 0.2674; 0.2144) |
Our calculations show very strong dependence of the structure (Figures 2 and 3) and elastic constants (Figure 4) on f(Si-O). The most interesting feature is that at higher ionicity degrees b-quartz (space group P3121) is not stable anymore, but transforms to a-quartz (space group P6222). It is likely that the real phase transition can be to some extent simulated by the transition with the ionicity degree change. The transformation is accompanied (Figure 2) by a volume gap (therefore it's the 1-order transition), though there is not any energy gap (Figure 1). The volume gap (+2,6%) is due to Si-O-Si angle gap without any abrupt change in bond lengths (Figure 3) and tetrahedral angles. Thus, in this transition SiO4-tetrahedra behave as rigid units. In accord with experiment our calculations predict b-quartz to be much more rigid (less compressible) than a-quartz (Figure 4).
In summary, a-b transition of quartz seems to be of the 1-st order, what is in agreement with the recent experimental evidences. This transition can be adequately represented by the rigid-unit mode model [4].
We acknowledge the financial support of RFFI (96-05-64567)
and ISSEP (s97-2849, p97-349).
1. V.S.Urusov, N.N.Eremin, A.R.Oganov (1998). Crystallography
Reports, 1998, v.43, N 5.
2. V.S.Urusov, N.N.Eremin (1997). Phys. Chem.
Miner., 1997, v.24, p.374-383.
3. G.Will, M.Bellotto,W.Parrish, M.Hart (1988). J.Appl.
Crystallogr., 1988, v.21, p.182-191.
4. M.T.Dove. Amer. Miner., 1997, v.82,
Iss 3-4, p. 213-244.
Fig. 1
Fig. 2
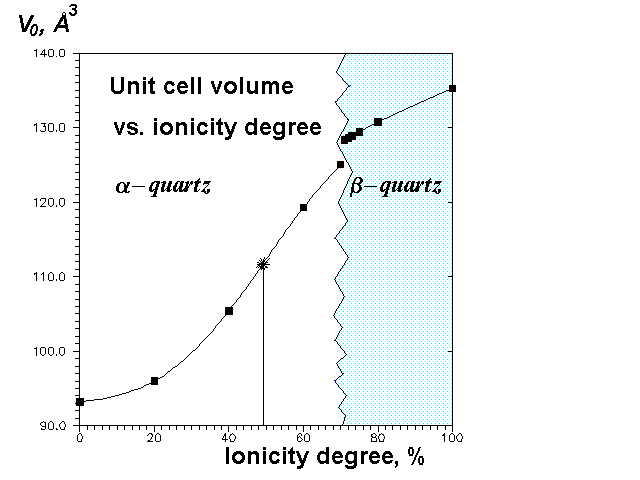
Fig. 3
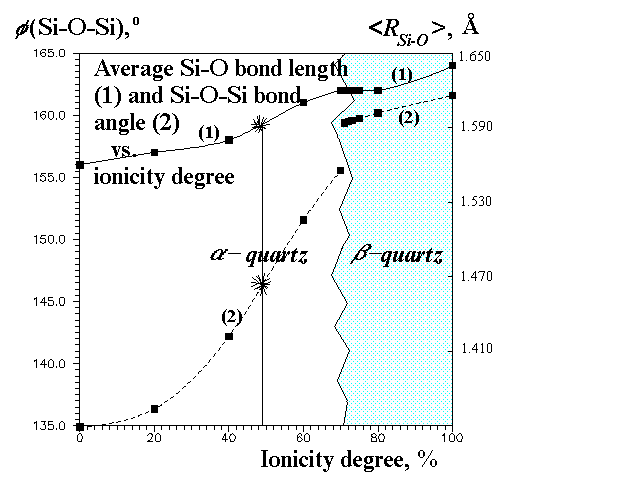
Fig. 4